Featured Core Publication:
Wurl JA, Mac Nair CE, Dietz JA, Shestopalov VI, Nickells RW. Contralateral Astrocyte Response to Acute Optic Nerve Damage Is Mitigated by PANX1 Channel Activity. Int J Mol Sci. 2023 Oct 27;24(21):15641. doi: 10.3390/ijms242115641. PMID: 37958624; PMCID: PMC10647301.
Glial reactivity is considered a hallmark of damage-induced innate immune responses in the central nervous system. In the visual system, unilateral optic nerve damage elicits dramatic glial reactivity in the retina directly affected by the lesion and a similar, albeit more modest, effect in the contralateral eye. Evaluation of astrocyte changes in a mouse model of optic nerve crush indicates that astrocyte reactivity, as a function of retinal coverage and cellular hypertrophy, occurs within both the experimental and contralateral retinas, although the hypertrophic response of the astrocytes in the contralateral eyes is delayed for at least 24 h. Evaluation of astrocytic reactivity as a function of Gfap expression indicates a similar, muted but significant, response in contralateral eyes. This constrained glial response is completely negated by conditional knock out of Panx1 in both astrocytes and Müller cells. Further studies are required to identify if this is an autocrine or a paracrine suppression of astroglial reactivity.
Figure 3 (click on image to enlarge)
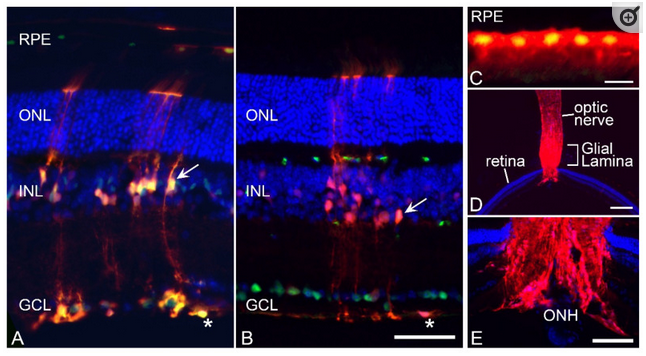
Validation of CRE activity in retinal macroglia. To confirm that Gfap-Cre transgenic mice successfully modified gene expression in target cells of the retina, this line was crossed with Rosa26-(LoxP)-tdTomato reporter mice and analyzed in frozen sections. (A) Section showing tdTomato fluorescence counterstained with an antibody against SOX9 which labels both astrocytes (asterisk) and Müller cells (arrow) and subsets of retinal pigmented epithelium (RPE) cells. The tdTomato transgene is expressed in the entire astrocyte population and a subset of Müller cells. (B) Section showing tdTomato fluorescence counterstained with an antibody against BRN3A, which stains retinal ganglion cells (RGCs). RGCs are negative for tdTomato expression and expression in the astrocytes (asterisk) and Müller cell (arrow) end feet are clearly visible adjacent to the ganglion cell layer (GCL). Scale bar (A,B) = 100 µm. (C) Higher magnification of a section of the RPE layer showing a cluster of tdTomato-expressing cells (counterstained with anti-SOX9). Scale bar = 20 µm. (D,E) low and higher magnification images of the optic nerve and optic nerve head (ONH) showing robust staining in the glial lamina. Scale bar (D) = 750 µm. Scale bar (E) = 300 µm. All sections were counter-stained with DAPI. Outer nuclear layer (ONL). Inner nuclear layer (INL).